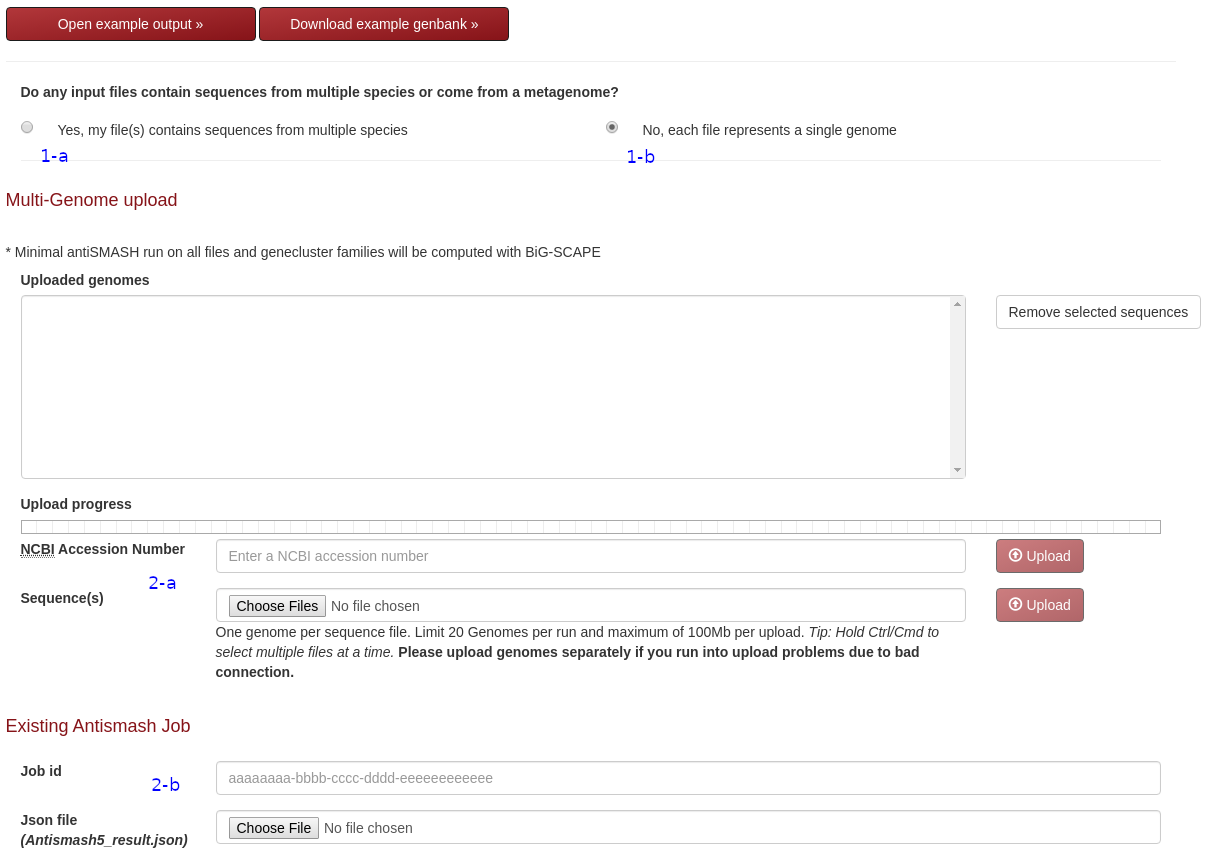
Mode Selection
- Select this option if the input file contains genomic information from multiple species e.g. Metagenome assemblies
- Select this option if the input file only contains genomic information from a specific organism
Input Genome
- Upload here if no antiSMASH annotation is present in file. Select a file or files to upload and click on the upload button. A minimal antiSMASH search will be performed automatically. Note: Multi-record fasta files will be processed as separate scaffolds. Input NCBI accession number or upload FASTA, Genbank or EMBL files
- Job id for genome previously run on public antiSMASH server (antismash.secondarymetabolites.org) or Upload file from finished antiSMASH (ex: [filename].gbk). Genbank files with cluster annotation can also be submitted here (see http://mibig.secondarymetabolites.org/ for example genbank cluster annotation format)

Email and Reference selection
- Email address to send job id link (optional). Be sure to bookmark result page if absent or select "save link in cookies" to display in results page.
- Reference dataset for Phylogeney and duplication analysis. Currently all phyla are available with some of them grouped together (See group1, 2 and 3 in reference set option). Download organism list
Run Options (Enable / Disable analysis)
- BGC proximity and duplication is always preformed
- Phylogeny may be disabled, if query is not in reference phyla this will save unnecessary load time
- Resistance models include Resfams and curated models from known drug targets
- Pfam DUF search highlights unknown reactions in BGCs
- Save run will store runid link in results page until next time browser's cookies are cleared. Default is set for private
Hmmer thresholds for hits - using model specific values for trusted cutoff and noise cutoff (default: trusted)
Custom model inputs
- Valid Hmmer file for resistance models. Model will be converted to hmmer v3
- Valid Hmmer file for core gene models. Model will be converted to hmmer v3
Accepted genbank formats require gene/CDS records for new job input while "cluster" annotations from antiSMASH are required when submitting into existing job section
Fasta formats are parsed through antiSMASH and nucleotide data is required for codon alignments in phylogeny analysis
Download example genbank file
Download example fasta file
Download custom hmm model file
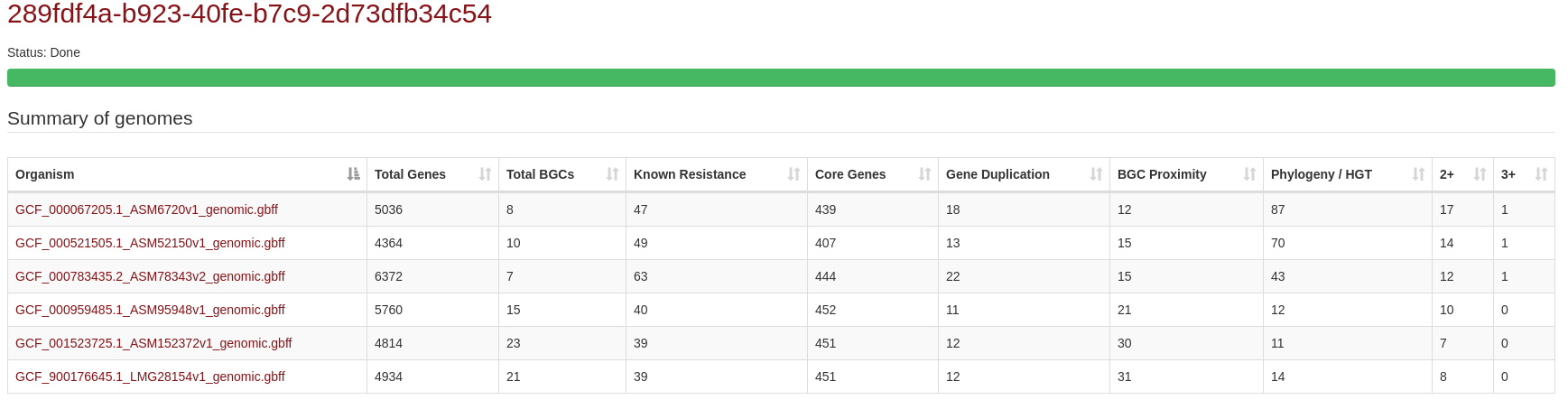
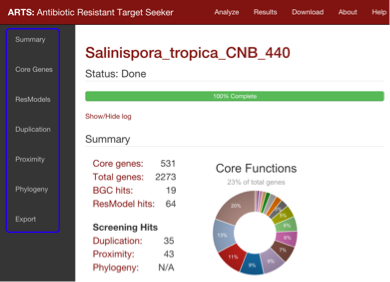
General layout of results summary:
The summaries of each individual ARTS run can be found in the first part of the results page. Results for
each genome can also be navigated through clicking on the files provided in the "Organism" column
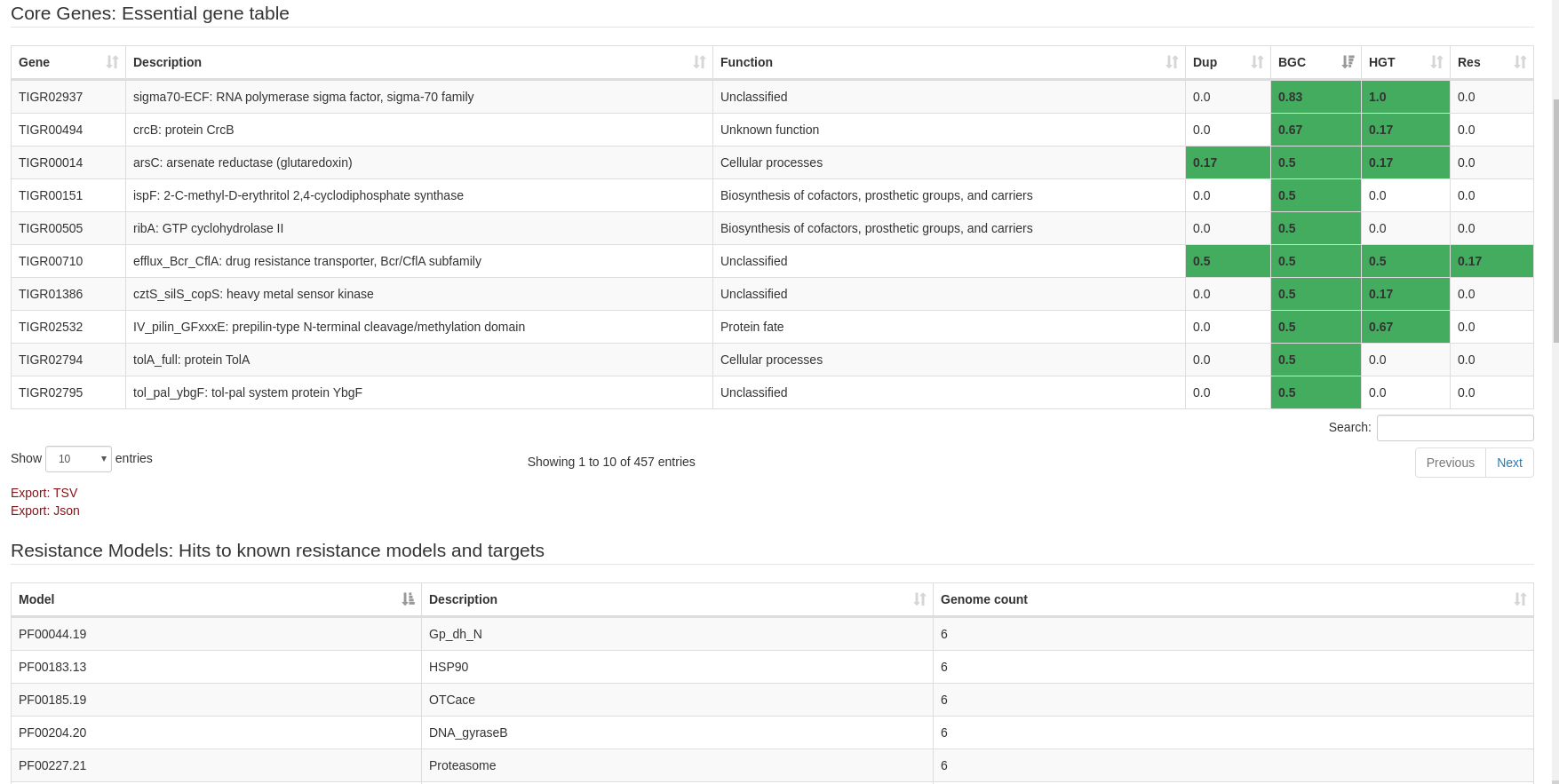
General datatables navigation:
Data representation style from multiple results are much like the same with individual ARTS run results (please see single results page example below).
One exception to mind is, in "Core Genes" table the numbers shown in different criterions represent the frequency of hits found
in total input number; "1.0" in all genomes, "0,0" in none. In "Resistance Models" and "Duplication" tables numbers
represent how many genomes have the corresponding hit.
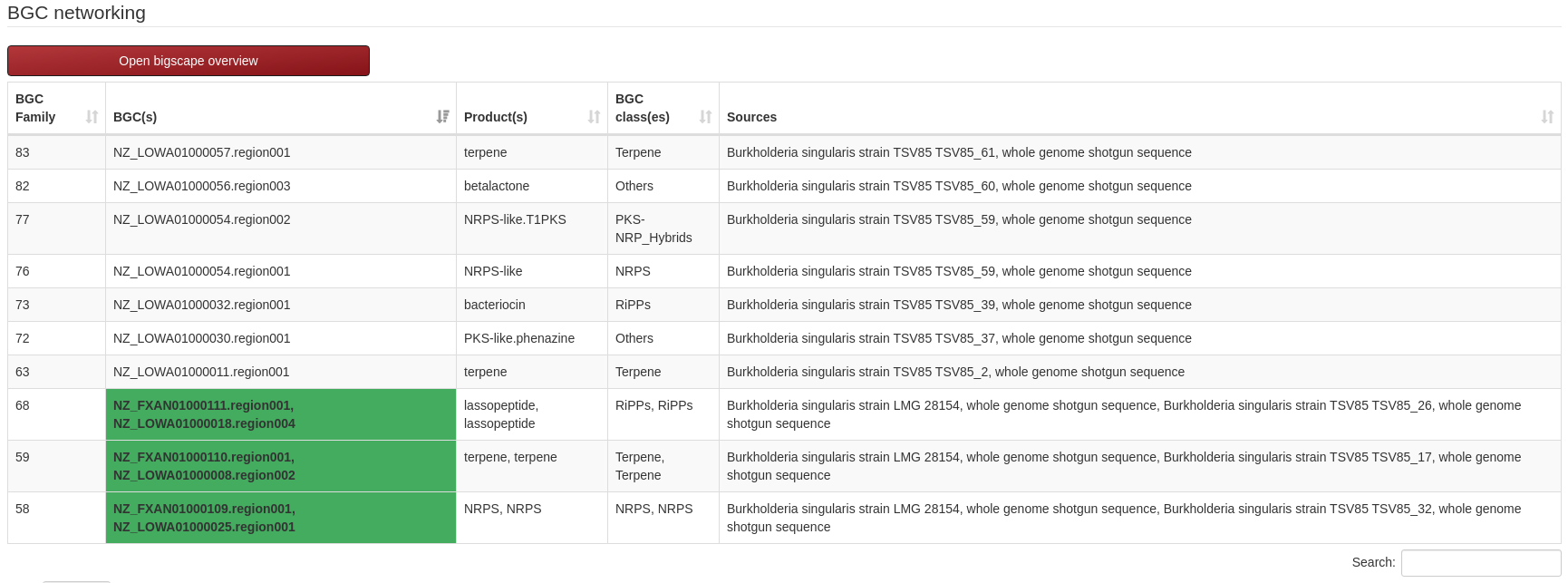
Bigscape overview:
Bigscape result of the given inputs are also represented at the bottom of the results page. Here, the clustered
BGCs from antiSMASH results are shown with associated core genes and resistance models. "Open bigscape overview"
option also allows visualization of the interactive bigscape result page of the clusters.
General layout & Interpreting results:
Results can be viewed from multiple perspectives represented by each section on the left navigation bar: Core genes, Resistance models, Duplication, BGC Proximity, and phylogeny.
The summary section gives counts for model hits and screening criteria as well as functional statistics of core gene hits.
Each section has an interactive table to explore the data with detailed visualization where applicable.
General datatables navigation:
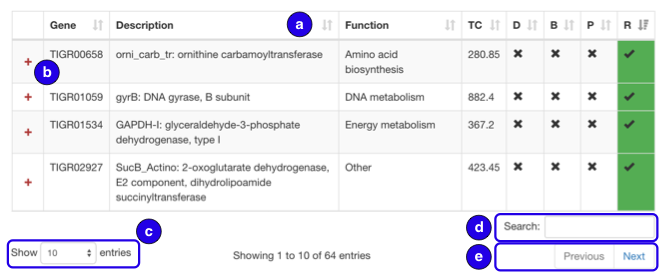
- Click table headers to sort ascending or descending
- Click to toggle expandable rows indicated with a plus/minus sign
- Select number of entries to view at a time
- Search to find rows with certain terms included
- Move to next / previous page of table
Core table:

- Quickly sort by clicking screening criteria titles
- D: Duplication, B: Biosynthetic Gene Cluster (BGC) proximity, P: Phylogeny, R: Resistance model hit
- Details for positive hits are provided for each in the expanded column and corresponding sections
Core table expanded row:
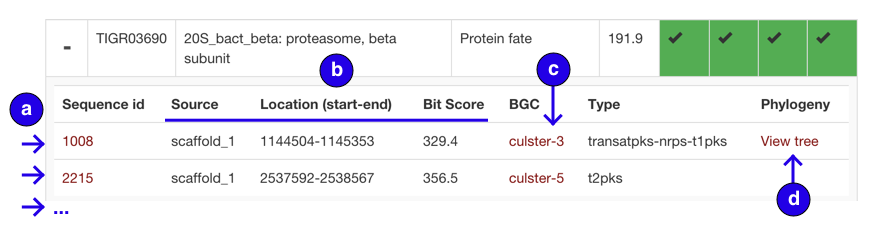
- Rows of sequence hits. Ids are assigned arbitrarily, roll over id to see original record description
- Metadata for location in genome and bitscore of model match
- BGC proximity hits. Click to open row in proximity section
- Phylogeny hit, click to view tree in phylogeny section
Duplicate table expanded row:
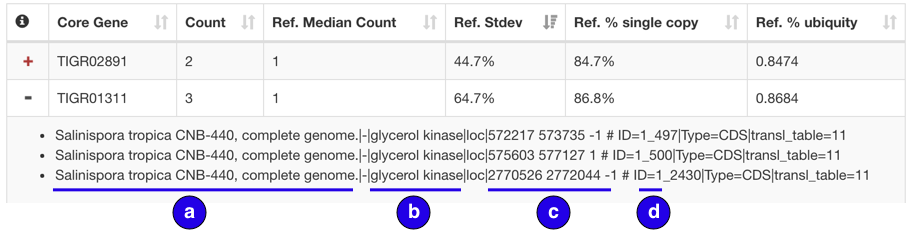
Fasta headers for duplication hits are listed in the expanded row with the following information
- Source of sequence / Organism
- Extracted gene information (Gene identifier if applicable and description)
- Location start, end, and strand
- Scaffold number
Proximity table expanded row:
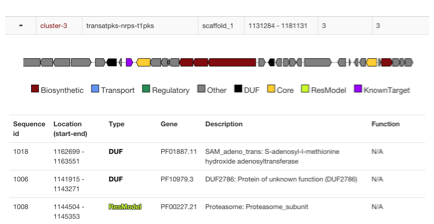
- Cluster id link. Click link to open in original antismash view
- Quick preview with overlay of hits. Click domains for popup of details
- Hit table gives type, description and function of model if applicable
Phylogeny view:
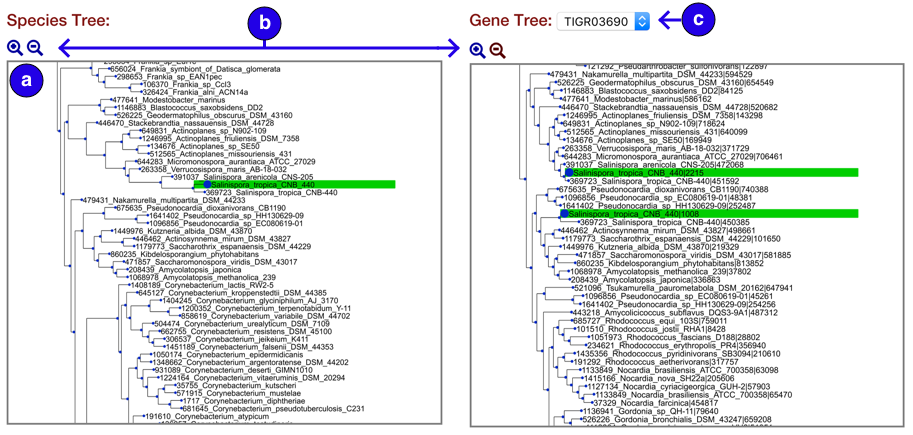
- Tree pane viewer for species and gene trees
- Zoom buttons to resize tree image
- Gene tree dropdown to select phylogeny hits directly
View example output