ARTS version 2 is now capable of:
- Analyzing all bacterial kingdom as well as metagenomic data (Please see reference sets in analyze page)
- Performing multi genome comparative analysis
- Performing BigSCAPE analysis on all found antismash clusters from multiple organisms
- Using clusters from antiSMASH v5 json formatted result file
- 30.05.2023 - Added option for exporting all results (compressed)
- 31.05.2023 - Updated BiG-SCAPE (v1.1.5)
- 07.11.2023 - Python3 beta version of ARTS is released on Docker Hub. You can find more detail here.
- 20.02.2024 - Python 3 version of ARTS is published on Github. You can find more detail here.
The goals of this tool are to automate the process of performing target direct genome mining1,2,3, search for potential novel antibiotic targets, and prioritize putative secondary metabolite gene clusters. The following initial automated steps, and associated genome mining tools, that are used to realize these goals are:
- Predict Secondary Metabolite gene clusters: Antibiotics & Secondary Metabolite Analysis SHell (antiSMASH)
- Identify known antibiotic targets & Domains of Unknown Function: Pfam models of essential genes known to be targeted from published work & DUF domains
- Identify known resistance factors: ResFams, manually curated models that include proteins from The Comprehensive Antibiotic Resistance Database (CARD), The LACtamase Engineering Database (LACED), and The Jacobi and Bush Collection
- Identify essential genes: ARTS comparative pipeline + TIGRfam Equivologs
Upon completion, the ARTS workflow uses these results to:
- Proximity check: Cross reference locations with Secondary Metabolite gene clusters
- Uncommon duplication check: Highlight potential repurposed primary metabolism genes
- Phylogenetic incongruence: Highlight essential genes with evidence of inter-genus horizontal transfer
- Visualize results: Provide an interactive format for rapid manual confirmation
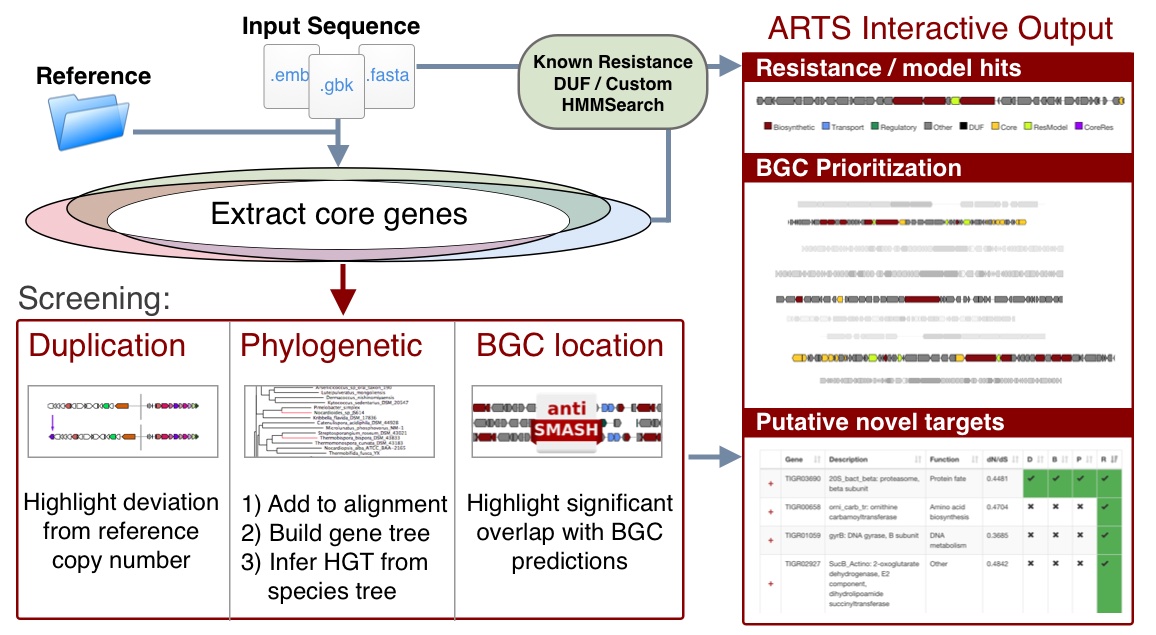
Potential novel targets are highlighted by exploring the space of essential genes in a query genome and filtering them by criteria associated with antibiotic resistance:
- Co-expression with secondary metabolite gene cluster: Self-resistance method to avoid suicide during production
- Duplication: To maintain non-resistant, likely higher fitness, version of gene
- Horizontal transfer: Acquisition of foreign version suggests significant advantage despite pressure to maintain species optimized version
Reference workflow:
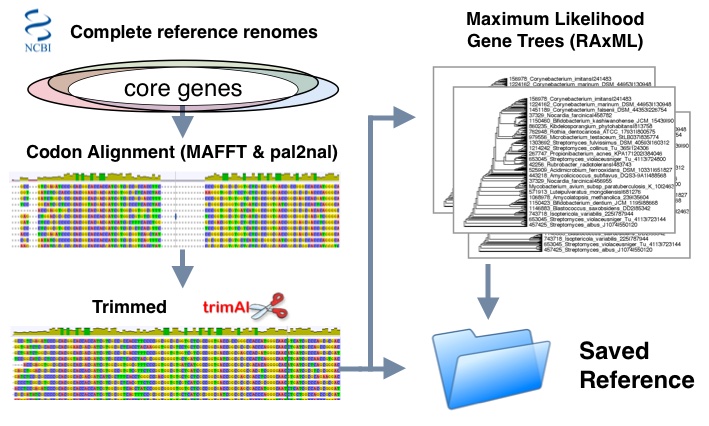
- Set of phyla specific complete reference genomes are used to determine core genes based on ubiquity
- Genes are classified by protein families and sorted into corresponding protein alignments using MAFFT lins-i method
- protein alignments and nucleotide sequences are run with pal2nal to produce codon alignments
- Trimming is done using trimal to optimize maximum likelihood estimations using "automated1" setting
- Final evolutionary trees are inferred in RaxML using General Time Reversible (GTR) substitution model and GAMMA distribution of site rate variation
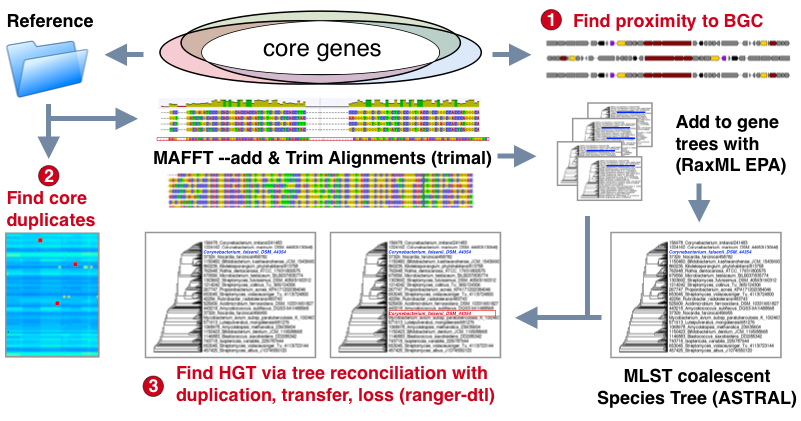
Query genome workflow:
- Biosynthetic Gene Clusters (BGCs) are identified with antiSMASH if this is not already present. (minimal run omitting extra options is performed)
- Known resistance & target models are searched and identified
- Core genome models from reference are used to identify and extract query genes.
- Duplications are marked based on deviation from the sum of the reference median count and standard deviation. Note: Draft genomes with repeat genes due to mis-assembly should be manually confirmed
- Sequences are added to corresponding reference alignments which with MAFFT --add which are used to place them on reference trees using RaxML's Evolutionary Placement Algorithm (EPA)
- A species tree is constructed by selecting all available single copy core genes with avg dn/ds < 1 and using ASTRAL to produce a coalescent tree from these gene trees
- A species tree is constructed by selecting all available single copy core genes with avg dn/ds < 1 and using ASTRAL to produce a coalescent tree from these gene trees
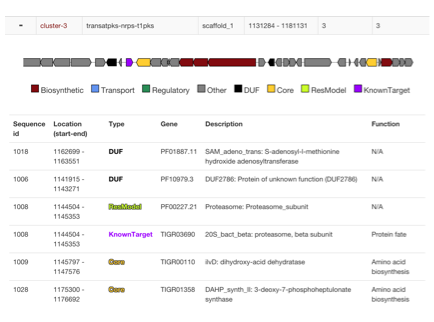
- Biosynthetic Gene Clusters (BGCs) are identified with antiSMASH or read from genbank "cluster" annotations
- Locations of Core genes, DUF, Resistance models and custom models are checked if they are within cluster boundaries
- Additional cluster visualization is presented to identify where hits are present and in what context
If you found ARTS to be helpful please cite the following publications:https://doi.org/10.1093/nar/gkaa374
Mungan,M.D., Alanjary,M., Blin,K., Weber,T., Medema,M.H. and Ziemert,N. (2020) ARTS 2.0: feature updates and expansion of the Antibiotic Resistant Target Seeker for comparative genome mining. Nucleic Acids Res.,10.1093/nar/gkaa374
Alanjary,M., Kronmiller,B., Adamek,M., Blin,K., Weber,T., Huson,D., Philmus,B. and Ziemert,N. (2017) The Antibiotic Resistant Target Seeker (ARTS), an exploration engine for antibiotic cluster prioritization and novel drug target discovery. Nucleic Acids Res.,10.1093/nar/gkx360
- Thaker, M. N., Wang, W., Spanogiannopoulos, P., Waglechner, N., King, A. M., Medina, R., & Wright, G. D. (2013). Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nature Biotechnology, 31(10), 922–927.
- Tang, X., Li, J., Millán-Aguiñaga, N., Zhang, J. J., O’Neill, E. C., Ugalde, J. A., … Moore, B. S. (2015). Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-directed Genome Mining. ACS Chemical Biology, 10(12), 2841–2849.
- Johnston, C. W., Skinnider, M. A., Dejong, C. A., Rees, P. N., Chen, G. M., Walker, C. G., … Magarvey, N. A. (2016). Assembly and clustering of natural antibiotics guides target identification. Nature Chemical Biology, 12(4), 233–239.
- Medema, M. H., Blin, K., Cimermancic, P., De Jager, V., Zakrzewski, P., Fischbach, M. A., … Breitling, R. (2011). AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Research, 39(SUPPL. 2), 339–346.